
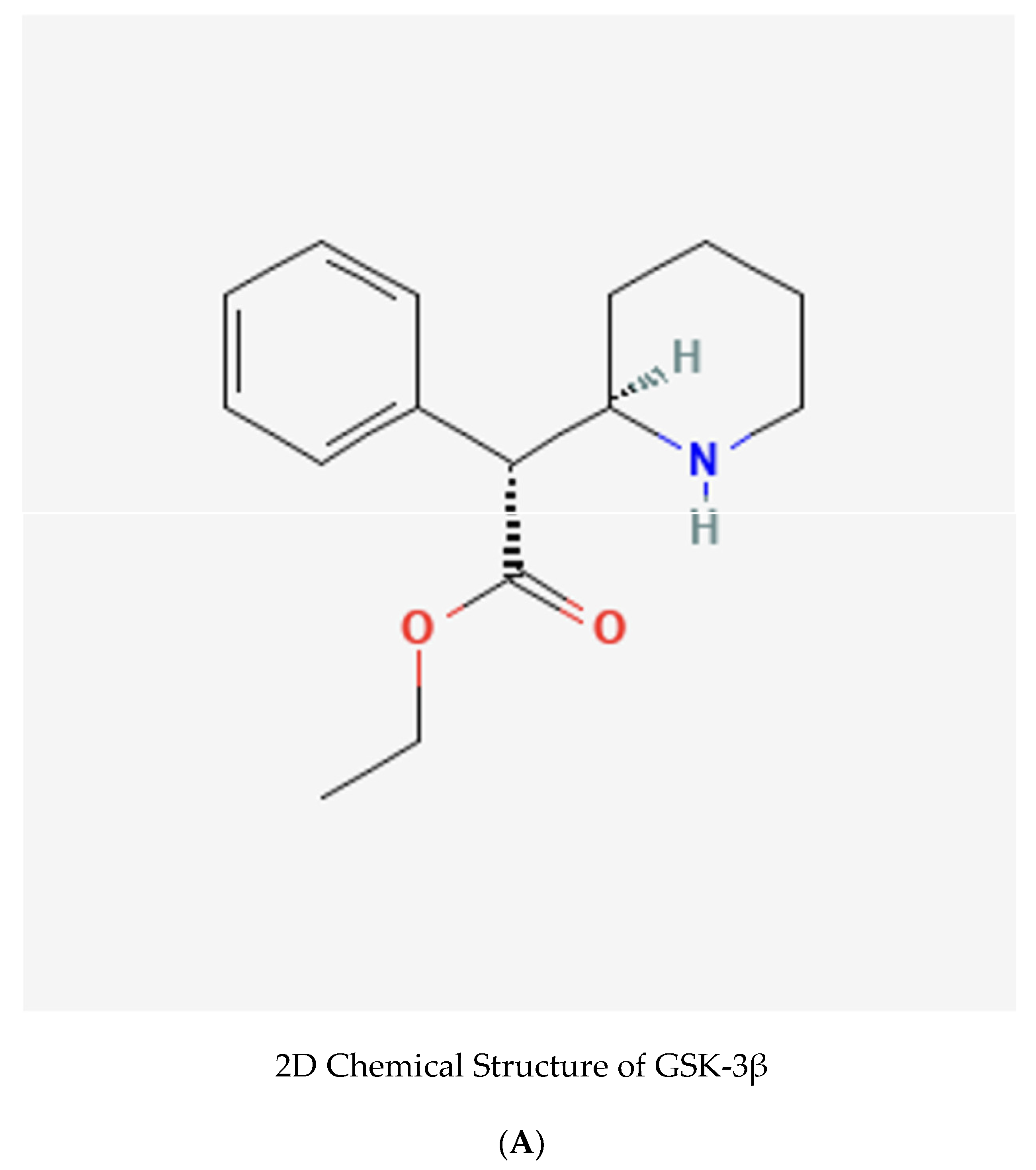
Beyond Amyloid: A Machine Learning-Driven Approach Reveals Properties of Potent GSK-3β Inhibitors Targeting Neurofibrillary Tangles


Abstract
:1. Introduction
2. Results
2.1. Strengths and Limitations of Individual and Majority Voting Ensemble Models
2.2. Important Features
3. Discussion
4. Materials and Methods
4.1. Obtaining the Dataset
4.2. Generating Descriptors of Chemical Compounds
4.3. Machine Learning Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bomasang-Layno, E.; Bronsther, R. Diagnosis and Treatment of Alzheimer’s Disease: An Update. Dela J. Public Health 2021, 7, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Nwadiugwu, M. Early-Onset Dementia: Key Issues Using a Relationship-Centred Care Approach. Postgrad. Med. J. 2021, 97, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Nwadiugwu, M.; Shen, H.; Deng, H.-W. Potential Molecular Mechanisms of Alzheimer’s Disease from Genetic Studies. Biology 2023, 12, 602. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell, Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.; Zhou, Y.; Park, C.; Yokozawa, T.; Jung, H.; Choi, J. Rosmarinic Acid Derivatives’ Inhibition of Glycogen Synthase Kinase-3β Is the Pharmacological Basis of Kangen-Karyu in Alzheimer’s Disease. Molecules 2018, 23, 2919. [Google Scholar] [CrossRef] [PubMed]
- Tse, K.; Cheng, A.; Ma, F.; Herrup, K. DNA Damage-associated Oligodendrocyte Degeneration Precedes Amyloid Pathology and Contributes to Alzheimer’s Disease and Dementia. Alzheimer’s Dement. 2018, 14, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Noguchi, K.; Michel, G.; Mercken, M.; Hoshi, M.; Ishiguro, K.; Imahori, K. Exposure of Rat Hippocampal Neurons to Amyloid β Peptide (25–35) Induces the Inactivation of Phosphatidyl Inositol-3 Kinase and the Activation of Tau Protein Kinase I/Glycogen Synthase Kinase-3β. Neurosci. Lett. 1996, 203, 33–36. [Google Scholar] [CrossRef]
- Leroy, K.; Boutajangout, A.; Authelet, M.; Woodgett, J.R.; Anderton, B.H.; Brion, J.P. The Active Form of Glycogen Synthase Kinase-3? Is Associated with Granulovacuolar Degeneration in Neurons in Alzheimer’s Disease. Acta Neuropathol. 2002, 103, 91–99. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau Protein Kinases: Involvement in Alzheimer’s Disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Nakayama, S.; Yumimoto, K.; Kawamura, A.; Nakayama, K.I. Degradation of the Endoplasmic Reticulum–Anchored Transcription Factor MyRF by the Ubiquitin Ligase SCFFbxw7 in a Manner Dependent on the Kinase GSK-3. J. Biol. Chem. 2018, 293, 5705–5714. [Google Scholar] [CrossRef] [PubMed]
- Gorris, R.; Fischer, J.; Erwes, K.L.; Kesavan, J.; Peterson, D.A.; Alexander, M.; Nöthen, M.M.; Peitz, M.; Quandel, T.; Karus, M.; et al. Pluripotent Stem Cell-Derived Radial Glia-like Cells as Stable Intermediate for Efficient Generation of Human Oligodendrocytes. Glia 2015, 63, 2152–2167. [Google Scholar] [CrossRef] [PubMed]
- Muyllaert, D.; Kremer, A.; Jaworski, T.; Borghgraef, P.; Devijver, H.; Croes, S.; Dewachter, I.; Van Leuven, F. Glycogen Synthase Kinase-3β, or a Link between Amyloid and Tau Pathology? Genes. Brain Behav. 2008, 7, 57–66. [Google Scholar] [CrossRef] [PubMed]
- de Barreda, E.G.; Pérez, M.; Ramos, P.G.; de Cristobal, J.; Martín-Maestro, P.; Morán, A.; Dawson, H.N.; Vitek, M.P.; Lucas, J.J.; Hernández, F.; et al. Tau-Knockout Mice Show Reduced GSK3-Induced Hippocampal Degeneration and Learning Deficits. Neurobiol. Dis. 2010, 37, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Jewett BE, T.B. Physiology, NMDA Receptor; StatPearls Publishing: St. Petersburg, FL, USA,, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519495/ (accessed on 23 November 2023).
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Tan, W.; Cao, X.; Wang, J.; Lv, H.; Wu, B.; Ma, H. Protective Effects of Lithium Treatment for Spatial Memory Deficits Induced by Tau Hyperphosphorylation in Splenectomized Rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1010–1015. [Google Scholar] [CrossRef]
- Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive Ensemble in QSAR Prediction for Drug Discovery. BMC Bioinform. 2019, 20, 521. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Cramer, R.D. The Inevitable QSAR Renaissance. J. Comput. Aided Mol. Des. 2012, 26, 35–38. [Google Scholar] [CrossRef]
- Karpov, P.; Godin, G.; Tetko, I.V. Transformer-CNN: Swiss Knife for QSAR Modeling and Interpretation. J. Cheminform 2020, 12, 17. [Google Scholar] [CrossRef]
- Jaganathan, K.; Tayara, H.; Chong, K.T. An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors. Pharmaceutics 2022, 14, 832. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, A.; Pérez-Castillo, Y.; Schürer, S.C.; Nicolotti, O.; Mangiatordi, G.F.; Borges, F.; Cordeiro, M.N.D.S.; Tejera, E.; Medina-Franco, J.L.; Cruz-Monteagudo, M. From Flamingo Dance to (Desirable) Drug Discovery: A Nature-Inspired Approach. Drug Discov. Today 2017, 22, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Maura C Kibbey Pharmacopeial Forum. Volume 36, 2010. Available online: www.drugfuture.com/Pharmacopoeia/usp38/data/v38332/usp38nf33s2_c1034.html#usp38nf33s2_c1034 (accessed on 23 November 2023).
- Berrouet, C.; Dorilas, N.; Rejniak, K.A.; Tuncer, N. Comparison of Drug Inhibitory Effects ([Formula: See Text]) in Monolayer and Spheroid Cultures. Bull. Math. Biol. 2020, 82, 68. [Google Scholar] [CrossRef] [PubMed]
- Więckowska, B.; Kubiak, K.B.; Jóźwiak, P.; Moryson, W.; Stawińska-Witoszyńska, B. Cohen’s Kappa Coefficient as a Measure to Assess Classification Improvement Following the Addition of a New Marker to a Regression Model. Int. J. Env. Res. Public Health 2022, 19, 10213. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Tanaka, K.; Kotera, M.; Funatsu, K. Comparison and Improvement of the Predictability and Interpretability with Ensemble Learning Models in QSPR Applications. J. Cheminform 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Kora, R. A Comprehensive Review on Ensemble Deep Learning: Opportunities and Challenges. J. King Saud. Univ.-Comput. Inf. Sci. 2023, 35, 757–774. [Google Scholar] [CrossRef]
- Ju, C.; Bibaut, A.; van der Laan, M. The Relative Performance of Ensemble Methods with Deep Convolutional Neural Networks for Image Classification. J. Appl. Stat. 2018, 45, 2800–2818. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.; Shanto, M.S.I.; Kabir, M.M.; Rahman, M.S.; Mridha, M.F. Heart Disease Prediction and Analysis Using Ensemble Architecture. In Proceedings of the 2022 International Conference on Decision Aid Sciences and Applications (DASA), Chiangrai, Thailand, 23–25 March 2022; IEEE: Piscataway, NJ, USA, 2022; pp. 1386–1390. [Google Scholar]
- Mahajan, P.; Uddin, S.; Hajati, F.; Moni, M.A. Ensemble Learning for Disease Prediction: A Review. Healthcare 2023, 11, 1808. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Haque, I.; Lu, H.; Moni, M.A.; Gide, E. Comparative Performance Analysis of K-Nearest Neighbour (KNN) Algorithm and Its Different Variants for Disease Prediction. Sci. Rep. 2022, 12, 6256. [Google Scholar] [CrossRef]
- Hassija, V.; Chamola, V.; Mahapatra, A.; Singal, A.; Goel, D.; Huang, K.; Scardapane, S.; Spinelli, I.; Mahmud, M.; Hussain, A. Interpreting Black-Box Models: A Review on Explainable Artificial Intelligence. Cogn. Comput. 2024, 16, 45–74. [Google Scholar] [CrossRef]
- Nambiar, A.; Harikrishnaa, S.; Sharanprasath, S. Model-Agnostic Explainable Artificial Intelligence Tools for Severity Prediction and Symptom Analysis on Indian COVID-19 Data. Front. Artif. Intell. 2023, 6, 1272506. [Google Scholar] [CrossRef]
- Garrido, J.J.; Simón, D.; Varea, O.; Wandosell, F. GSK3 Alpha and GSK3 Beta Are Necessary for Axon Formation. FEBS Lett. 2007, 581, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Garrido, J.J.; Wandosell, F.G. Modulation of GSK-3 as a Therapeutic Strategy on Tau Pathologies. Front. Mol. Neurosci. 2011, 4, 24. [Google Scholar] [CrossRef]
- Davies, M.P.; Benitez, R.; Perez, C.; Jakupovic, S.; Welsby, P.; Rzepecka, K.; Alder, J.; Davidson, C.; Martinez, A.; Hayes, J.M. Structure-Based Design of Potent Selective Nanomolar Type-II Inhibitors of Glycogen Synthase Kinase-3β. J. Med. Chem. 2021, 64, 1497–1509. [Google Scholar] [CrossRef]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef]
- Arfeen, M.; Bharatam, P. Design of Glycogen Synthase Kinase-3 Inhibitors: An Overview on Recent Advancements. Curr. Pharm. Des. 2013, 19, 4755–4775. [Google Scholar] [CrossRef]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Derivation and Applications of Molecular Descriptors Based on Approximate Surface Area. In Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery; Humana Press: Totowa, NJ, USA, 2004; pp. 261–278. [Google Scholar]
- Yeung, W.; Ruan, Z.; Kannan, N. Emerging Roles of the AC-Β4 Loop in Protein Kinase Structure, Function, Evolution, and Disease. IUBMB Life 2020, 72, 1189–1202. [Google Scholar] [CrossRef]
- Crippen, G.M. VRI: 3D QSAR at Variable Resolution. J. Comput. Chem. 1999, 20, 1577–1585. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A Public Information System for Analyzing Bioactivities of Small Molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org (accessed on 23 November 2023).
- Kaneko, H. Molecular Descriptors, Structure Generation, and Inverse QSAR/QSPR Based on SELFIES. ACS Omega 2023, 8, 21781–21786. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Dogan, A.; Birant, D. A Weighted Majority Voting Ensemble Approach for Classification. In Proceedings of the 2019 4th International Conference on Computer Science and Engineering (UBMK), Samsun, Turkey, 1–15 September 2019; IEEE: Piscataway, NJ, USA, 2019; pp. 1–6. [Google Scholar]
- Krstajic, D.; Buturovic, L.J.; Leahy, D.E.; Thomas, S. Cross-Validation Pitfalls When Selecting and Assessing Regression and Classification Models. J. Cheminform 2014, 6, 10. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | SVM | XGB | NN | |||||||||||||
| Training | Validation | Test | New (EVD) | Training | Validation | Test | New (EVD) | Training | Validation | Test | New (EVD) | |||||
| Recall | 0.87 | 0.87 | 0.88 | 0.67 | 1 | 0.86 | 0.91 | 0.68 | 0.73 | 0.92 | 0.91 | 0.7 | ||||
| Accuracy | 0.93 | 0.9 | 0.9 | 0.74 | 1 | 0.84 | 0.88 | 0.69 | 0.76 | 0.93 | 0.94 | 0.72 | ||||
| AUC–ROC | 0.93 | 0.91 | 0.9 | 0.72 | 1 | 0.84 | 0.88 | 0.69 | 0.76 | 0.97 | 0.96 | 0.79 | ||||
| F1-Score | 0.92 | 0.91 | 0.9 | 0.55 | 1 | 0.85 | 0.52 | 0.5 | 0.75 | 0.94 | 0.94 | 0.54 | ||||
| Cohen’s Kappa | 0.86 | 0.81 | 0.79 | 0.38 | 1 | 0.69 | 0.76 | 0.3 | 0.52 | 0.87 | 0.88 | 0.35 | ||||
| Ensemble | LogR | KNN | RF | |||||||||||||
| Training | Validation | Test | New (EVD) | Training | Validation | Test | New (EVD) | Training | Validation | Test | New (EVD) | Training | Validation | Test | New (EVD) | |
| 0.97 | 0.93 | 0.94 | 0.71 | 0.94 | 0.82 | 0.82 | 0.69 | 0.84 | 0.89 | 0.88 | 0.68 | 1 | 0.86 | 0.88 | 0.68 | |
| 0.97 | 0.87 | 0.87 | 0.71 | 0.95 | 0.79 | 0.78 | 0.69 | 0.86 | 0.83 | 0.82 | 0.69 | 1 | 0.84 | 0.84 | 0.73 | |
| 0.97 | 0.87 | 0.86 | 0.71 | 0.95 | 0.78 | 0.78 | 0.69 | 0.86 | 0.83 | 0.82 | 0.69 | 1 | 0.84 | 0.84 | 0.72 | |
| 0.97 | 0.89 | 0.88 | 0.54 | 0.95 | 0.8 | 0.79 | 0.51 | 0.86 | 0.85 | 0.83 | 0.51 | 1 | 0.85 | 0.85 | 0.55 | |
| 0.94 | 0.75 | 0.74 | 0.35 | 0.72 | 0.57 | 0.56 | 0.31 | 0.72 | 0.66 | 0.65 | 0.3 | 1 | 0.67 | 0.68 | 0.37 |
| S/N | SVM | logR | RF | XGB |
|---|---|---|---|---|
| 1 | fr_phenol | BCUT2D_LOGPHI | MaxAbsEstateIndex | SMR_VSA3 |
| 2 | fr_phenol_noOrthoHbond | VSA_EState10 | BCUT2D_MRHI | Chi2v |
| 3 | frAr_OH | fr_unbrch_alkane | MaxEStateIndex | frAr_OH |
| 4 | fr_N_O | Estate_VSA2 | FpDensityMorgan2 | fr_methoxy |
| 5 | fr_priamide | fr_N_O | FpDensityMorgan3 | FpDensityMorgan3 |
| 6 | PEOE_VSA11 | fr_sulfonamd | PEOE_VSA6 | HeavyAtomMolWt |
| 7 | SlogP_VSA7 | fr_allylic_oxid | VSA_EState3 | MaxAbsEstateIndex |
| 8 | fr_imidazole | Estate_VSA6 | Chi2v | NumRotatableBonds |
| 9 | fr_piperdine | fr_Imine | MolWt | Chi4n |
| 10 | fr_C_S | SlogP_VSA3 | PEOE_VSA9 | PEOE_VSA13 |
| 11 | BCUT2D_MWLOW | PEOE_VSA12 | Chi1v | SMR_VSA7 |
| 12 | fr_methoxy | Avglpc | MolWt | Fr_para_hydroxylation |
| 13 | BCUT2D_CHGLO | fr_sulfide | PEOE_VSA9 | BCUT2D_MWLOW |
| 14 | PEOE_VSA3 | SlogP_VSA1 | Chi1v | MolMR |
| 15 | fr_sulfide | fr_imidazole | BCUT2D_MWHI | HallKierAlpha |
| 16 | lpc | fr_methoxy | ExactMolWt | MaxPartialCharge |
| 17 | Estate_VSA8 | SMR_VSA3 | Estate_VSA2 | VSA_Estate9 |
| 18 | Avglpc | fr_benzene | Kappa1 | Estate_VSA2 |
| 19 | fr_oxime | PEOE_VSA6 | SlogP_VSA1 | SMR_VSA1 |
| 20 | fr_hdrzone | fr_ether | Estate_VSA8 | fr_NHO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwadiugwu, M.; Onwuekwe, I.; Ezeanolue, E.; Deng, H. Beyond Amyloid: A Machine Learning-Driven Approach Reveals Properties of Potent GSK-3β Inhibitors Targeting Neurofibrillary Tangles. Int. J. Mol. Sci. 2024, 25, 2646. https://doi.org/10.3390/ijms25052646
Nwadiugwu M, Onwuekwe I, Ezeanolue E, Deng H. Beyond Amyloid: A Machine Learning-Driven Approach Reveals Properties of Potent GSK-3β Inhibitors Targeting Neurofibrillary Tangles. International Journal of Molecular Sciences. 2024; 25(5):2646. https://doi.org/10.3390/ijms25052646
Chicago/Turabian StyleNwadiugwu, Martin, Ikenna Onwuekwe, Echezona Ezeanolue, and Hongwen Deng. 2024. "Beyond Amyloid: A Machine Learning-Driven Approach Reveals Properties of Potent GSK-3β Inhibitors Targeting Neurofibrillary Tangles" International Journal of Molecular Sciences 25, no. 5: 2646. https://doi.org/10.3390/ijms25052646