

Neutrophil Extracellular Traps Upregulate p21 and Suppress Cell Cycle Progression to Impair Endothelial Regeneration after Inflammatory Lung Injury
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Patients
2.2. Animals
2.3. Endotoxemia Induction
2.4. Isolation of Neutrophils and NET Production
2.5. Quantification of dsDNA and MPO-DNA Complexes
2.6. Histopathological Analysis
2.7. Immunohistochemistry
2.8. Evans Blue Albumin (EBA) Tracer Measurement of Pulmonary Transvascular Permeability
2.9. EdU Assay of EC Proliferation
2.10. Intracellular EdU Flow Analysis
2.11. Cell Culture and Treatments
2.12. Cell Viability Assay
2.13. Immunofluorescence
2.14. Western Blot
2.15. Cell Cycle Analysis
2.16. Real-Time Quantitative PCR
2.17. RNA Sequencing (RNA-Seq)
2.18. Detection of Reactive Oxygen Species (ROS)
2.19. Mitochondrial Membrane Potential (MMP)
2.20. Statistical Analysis
3. Results
3.1. Increased NET Formation of Inflammatory Lung Injury Following Septic Endotoxemia
3.2. Degradation of NETs Attenuates Lung Inflammation and Facilitates Endothelial Regeneration
3.3. NETs Impair Endothelial Proliferation by Inducing Endothelial Cell Cycle Arrest
3.4. NETs Upregulate p21 and Inhibit Cell Cycle-Related Proteins
3.5. CDKN1A Knockdown Protects Endothelial Cells from NET-Induced Regeneration Defects
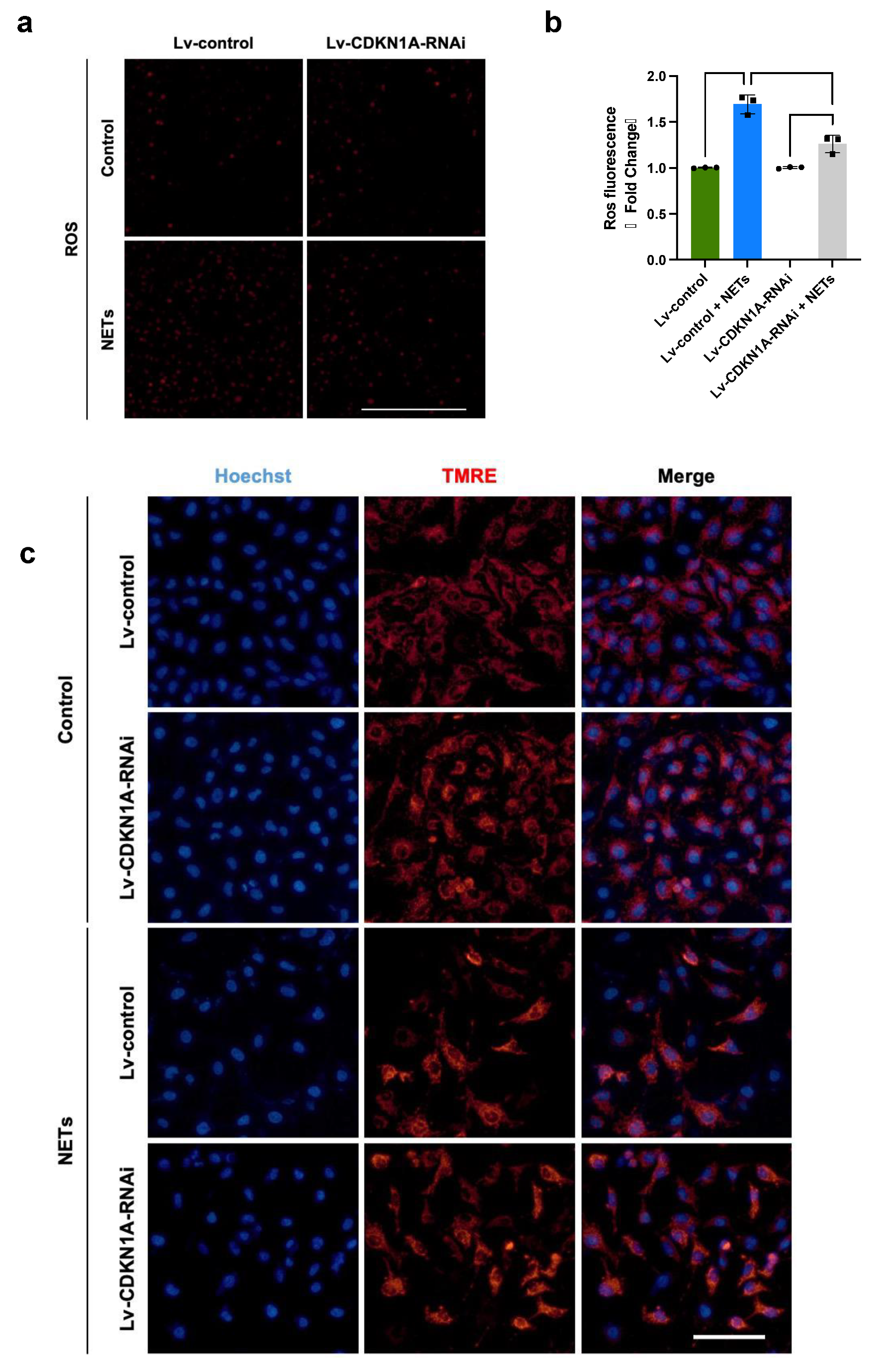
3.6. NETs Impair Mitochondrial Bioenergetics via p21/Cyclin B1/CDK1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Xiong, S.; Ye, Z.; Hong, Z.; Di, A.; Tsang, K.M.; Gao, X.; An, S.; Mittal, M.; Vogel, S.M.; et al. Caspase-11-Mediated Endothelial Pyroptosis Underlies Endotoxemia-Induced Lung Injury. J. Clin. Investig. 2017, 127, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e5. [Google Scholar] [CrossRef]
- Maniatis, N.A.; Orfanos, S.E. The Endothelium in Acute Lung Injury/Acute Respiratory Distress Syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Nature 2004, 303, 4. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, D.; Wang, Y.; Guo, K.; Spencer, C.B.; Ortoga, L.; Qu, M.; Shi, Y.; Shao, Y.; Wang, Z.; et al. METTL3-Mediated N6-Methyladenosine Exacerbates Ferroptosis via m6A-IGF2BP2-Dependent Mitochondrial Metabolic Reprogramming in Sepsis-Induced Acute Lung Injury. Clin. Transl. Med. 2023, 13, e1389. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Qu, M.; Li, W.; Wu, D.; Cata, J.P.; Miao, C. Neutrophil, Neutrophil Extracellular Traps and Endothelial Cell Dysfunction in Sepsis. Clin. Transl. Med. 2023, 13, e1170. [Google Scholar] [CrossRef]
- Zhu, S.; Yu, Y.; Qu, M.; Qiu, Z.; Zhang, H.; Miao, C.; Guo, K. Neutrophil Extracellular Traps Contribute to Immunothrombosis Formation via the STING Pathway in Sepsis-Associated Lung Injury. Cell Death Discov. 2023, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yu, Y.; Ren, Y.; Xu, L.; Wang, H.; Ling, X.; Jin, L.; Hu, Y.; Zhang, H.; Miao, C.; et al. The Emerging Roles of Neutrophil Extracellular Traps in Wound Healing. Cell Death Dis. 2021, 12, 984. [Google Scholar] [CrossRef] [PubMed]
- Safi, R.; Kallas, R.; Bardawil, T.; Mehanna, C.J.; Abbas, O.; Hamam, R.; Uthman, I.; Kibbi, A.-G.; Nassar, D. Neutrophils Contribute to Vasculitis by Increased Release of Neutrophil Extracellular Traps in Behçet’s Disease. J. Dermatol. Sci. 2018, 92, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, Y.; Zhang, Y.; Shen, D.; Wang, X.; Ge, R.; Zhang, M.; Xia, Y.; Wang, X. Antiphospholipid Antibody-Activated NETs Exacerbate Trophoblast and Endothelial Cell Injury in Obstetric Antiphospholipid Syndrome. J. Cell Mol. Med. 2020, 24, 6690–6703. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil Extracellular Traps Released by Neutrophils Impair Revascularization and Vascular Remodeling after Stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Marsboom, G.; Jambusaria, A.; Xiong, S.; Toth, P.T.; Benevolenskaya, E.V.; Rehman, J.; Malik, A.B. Sox17 Is Required for Endothelial Regeneration Following Inflammation-Induced Vascular Injury. Nat. Commun. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Yang, L.-Y.; Luo, Q.; Lu, L.; Zhu, W.-W.; Sun, H.-T.; Wei, R.; Lin, Z.-F.; Wang, X.-Y.; Wang, C.-Q.; Lu, M.; et al. Increased Neutrophil Extracellular Traps Promote Metastasis Potential of Hepatocellular Carcinoma via Provoking Tumorous Inflammatory Response. J. Hematol. Oncol. 2020, 13, 3. [Google Scholar] [CrossRef]
- Nie, M.; Yang, L.; Bi, X.; Wang, Y.; Sun, P.; Yang, H.; Liu, P.; Li, Z.; Xia, Y.; Jiang, W. Neutrophil Extracellular Traps Induced by IL8 Promote Diffuse Large B-Cell Lymphoma Progression via the TLR9 Signaling. Clin. Cancer Res. 2019, 25, 1867–1879. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, Z.; Zhu, C.; Ni, B.; Wang, S.; Yang, S.; Yu, F.; Zhao, E.; Li, Q.; Zhao, G. Neutrophil Extracellular Traps Promote Metastasis in Gastric Cancer Patients with Postoperative Abdominal Infectious Complications. Nat. Commun. 2022, 13, 1017. [Google Scholar] [CrossRef]
- Zu, G.; Yao, J.; Ji, A.; Ning, S.; Luo, F.; Li, Z.; Feng, D.; Rui, Y.; Li, Y.; Wang, G.; et al. Nurr1 Promotes Intestinal Regeneration after Ischemia/Reperfusion Injury by Inhibiting the Expression of P21 (Waf1/Cip1). J. Mol. Med. 2017, 95, 83–95. [Google Scholar] [CrossRef]
- Li, N.; Liu, B.; Xiong, R.; Li, G.; Wang, B.; Geng, Q. HDAC3 Deficiency Protects against Acute Lung Injury by Maintaining Epithelial Barrier Integrity through Preserving Mitochondrial Quality Control. Redox Biol. 2023, 63, 102746. [Google Scholar] [CrossRef]
- Akhter, M.Z.; Chandra Joshi, J.; Balaji Ragunathrao, V.A.; Maienschein-Cline, M.; Proia, R.L.; Malik, A.B.; Mehta, D. Programming to S1PR1+ Endothelial Cells Promotes Restoration of Vascular Integrity. Circ. Res. 2021, 129, 221–236. [Google Scholar] [CrossRef]
- Huang, X.; Dai, Z.; Cai, L.; Sun, K.; Cho, J.; Albertine, K.H.; Malik, A.B.; Schraufnagel, D.E.; Zhao, Y.-Y. Endothelial p110γPI3K Mediates Endothelial Regeneration and Vascular Repair After Inflammatory Vascular Injury. Circulation 2016, 133, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Weiner, A.I.; Neupauer, K.M.; de Mello Costa, M.F.; Palashikar, G.; Adams-Tzivelekidis, S.; Mangalmurti, N.S.; Vaughan, A.E. Regeneration of the Pulmonary Vascular Endothelium after Viral Pneumonia Requires COUP-TF2. Sci. Adv. 2020, 6, eabc4493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, T.; Jin, J.; Liu, Y.; Li, B.; Sun, Q.; Tian, J.; Zhao, H.; Liu, Z.; Ma, S.; et al. Interactions between Neutrophil Extracellular Traps and Activated Platelets Enhance Procoagulant Activity in Acute Stroke Patients with ICA Occlusion. EBioMedicine 2020, 53, 102671. [Google Scholar] [CrossRef] [PubMed]
- von Meijenfeldt, F.A.; Stravitz, R.T.; Zhang, J.; Adelmeijer, J.; Zen, Y.; Durkalski, V.; Lee, W.M.; Lisman, T. Generation of Neutrophil Extracellular Traps in Patients with Acute Liver Failure Is Associated with Poor Outcome. Hepatology 2022, 75, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Mestres-Arenas, A.; Fajas, L.; Leal-Esteban, L.C. The Multifaceted Role of Cell Cycle Regulators in the Coordination of Growth and Metabolism. FEBS J. 2021, 288, 3813–3833. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-Regulated Mitochondrial Bioenergetics in Cell Cycle Progression and Tumor Resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fan, M.; Candas, D.; Zhang, T.-Q.; Qin, L.; Eldridge, A.; Wachsmann-Hogiu, S.; Ahmed, K.M.; Chromy, B.A.; Nantajit, D.; et al. Cyclin B1/Cdk1 Coordinates Mitochondrial Respiration for Cell-Cycle G2/M Progression. Dev. Cell 2014, 29, 217–232. [Google Scholar] [CrossRef]
- Qin, L.; Fan, M.; Candas, D.; Jiang, G.; Papadopoulos, S.; Tian, L.; Woloschak, G.; Grdina, D.J.; Li, J.J. CDK1 Enhances Mitochondrial Bioenergetics for Radiation-Induced DNA Repair. Cell Rep. 2015, 13, 2056–2063. [Google Scholar] [CrossRef]
- Evans, C.E.; Iruela-Arispe, M.L.; Zhao, Y.-Y. Mechanisms of Endothelial Regeneration and Vascular Repair and Their Application to Regenerative Medicine. Am. J. Pathol. 2021, 191, 52–65. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Gao, X.-P.; Zhao, Y.D.; Mirza, M.K.; Frey, R.S.; Kalinichenko, V.V.; Wang, I.-C.; Costa, R.H.; Malik, A.B. Endothelial Cell-Restricted Disruption of FoxM1 Impairs Endothelial Repair Following LPS-Induced Vascular Injury. J. Clin. Investig. 2006, 116, 2333–2343. [Google Scholar] [CrossRef]
- Park, C.; Lee, T.-J.; Bhang, S.H.; Liu, F.; Nakamura, R.; Oladipupo, S.S.; Pitha-Rowe, I.; Capoccia, B.; Choi, H.S.; Kim, T.M.; et al. Injury-Mediated Vascular Regeneration Requires Endothelial ER71/ETV2. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 86–96. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, X.; Machireddy, N.; Evans, C.E.; Trewartha, S.D.; Hu, G.; Fang, Y.; Mutlu, G.M.; Wu, D.; Zhao, Y.-Y. Endothelial FoxM1 Reactivates Aging-Impaired Endothelial Regeneration for Vascular Repair and Resolution of Inflammatory Lung Injury. Sci. Transl. Med. 2023, 15, eabm5755. [Google Scholar] [CrossRef]
- Wang, M.; Yan, J.; He, X.; Zhong, Q.; Zhan, C.; Li, S. Candidate Genes and Pathogenesis Investigation for Sepsis-Related Acute Respiratory Distress Syndrome Based on Gene Expression Profile. Biol. Res. 2016, 49, 25. [Google Scholar] [CrossRef]
- Brühl, T.; Heeschen, C.; Aicher, A.; Jadidi, A.S.; Haendeler, J.; Hoffmann, J.; Schneider, M.D.; Zeiher, A.M.; Dimmeler, S.; Rössig, L. p21Cip1 Levels Differentially Regulate Turnover of Mature Endothelial Cells, Endothelial Progenitor Cells, and in Vivo Neovascularization. Circ. Res. 2004, 94, 686–692. [Google Scholar] [CrossRef]
- Abcejo, A.; Andrejko, K.M.; Ochroch, E.A.; Raj, N.R.; Deutschman, C.S. Impaired Hepatocellular Regeneration in Murine Sepsis Is Dependent on Regulatory Protein Levels. Shock 2011, 36, 471–477. [Google Scholar] [CrossRef]
- Yang, Q.-H.; Liu, D.-W.; Long, Y.; Liu, H.-Z.; Chai, W.-Z.; Wang, X.-T. Acute Renal Failure during Sepsis: Potential Role of Cell Cycle Regulation. J. Infect. 2009, 58, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, D.; Wang, X.; Yang, R.; Shi, Y.; Long, Y.; Liu, H.; He, H.; Zhou, X.; Tang, B. G1 Cell Cycle Arrest Signaling in Hepatic Injury after Intraperitoneal Sepsis in Rats. Inflamm. Res. 2011, 60, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-X.; Dong, Z.-J.; Deng, S.-X.; Tian, Y.-M.; Xiao, Y.-J.; Li, X.; Ma, X.-R.; Li, L.; Li, P.; Chang, H.-Z.; et al. GDF11 Slows Excitatory Neuronal Senescence and Brain Ageing by Repressing P21. Nat. Commun. 2023, 14, 7476. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and Consequences of Endothelial Cell Senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Billimoria, R.; Bhatt, P. Senescence in Cancer: Advances in Detection and Treatment Modalities. Biochem. Pharmacol. 2023, 215, 115739. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and Apoptosis: Dueling or Complementary Cell Fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-Alpha-Induced ROS Production Triggering Apoptosis Is Directly Linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and Cancer—Role and Therapeutic Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]
- Arias, C.F.; Ballesteros-Tato, A.; García, M.I.; Martín-Caballero, J.; Flores, J.M.; Martínez-A, C.; Balomenos, D. p21CIP1/WAF1 Controls Proliferation of Activated/Memory T Cells and Affects Homeostasis and Memory T Cell Responses. J. Immunol. 2007, 178, 2296–2306. [Google Scholar] [CrossRef]
- Trakala, M.; Arias, C.F.; García, M.I.; Moreno-Ortiz, M.C.; Tsilingiri, K.; Fernández, P.J.; Mellado, M.; Díaz-Meco, M.T.; Moscat, J.; Serrano, M.; et al. Regulation of Macrophage Activation and Septic Shock Susceptibility via P21(WAF1/CIP1). Eur. J. Immunol. 2009, 39, 810–819. [Google Scholar] [CrossRef]
- Rackov, G.; Hernández-Jiménez, E.; Shokri, R.; Carmona-Rodríguez, L.; Mañes, S.; Álvarez-Mon, M.; López-Collazo, E.; Martínez-A, C.; Balomenos, D. P21 Mediates Macrophage Reprogramming through Regulation of P50-P50 NF-κB and IFN-β. J. Clin. Investig. 2016, 126, 3089–3103. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101. [Google Scholar] [CrossRef]
- Huang, S.; Xu, M.; Liu, L.; Yang, J.; Wang, H.; Wan, C.; Deng, W.; Tang, Q. Autophagy Is Involved in the Protective Effect of P21 on LPS-Induced Cardiac Dysfunction. Cell Death Dis. 2020, 11, 554. [Google Scholar] [CrossRef]
- George, P.M.; Reed, A.; Desai, S.R.; Devaraj, A.; Faiez, T.S.; Laverty, S.; Kanwal, A.; Esneau, C.; Liu, M.K.C.; Kamal, F.; et al. A Persistent Neutrophil-Associated Immune Signature Characterizes Post-COVID-19 Pulmonary Sequelae. Sci. Transl. Med. 2022, 14, eabo5795. [Google Scholar] [CrossRef]
- Pisareva, E.; Badiou, S.; Mihalovičová, L.; Mirandola, A.; Pastor, B.; Kudriavtsev, A.; Berger, M.; Roubille, C.; Fesler, P.; Klouche, K.; et al. Persistence of Neutrophil Extracellular Traps and Anticardiolipin Auto-Antibodies in Post-Acute Phase COVID-19 Patients. J. Med. Virol. 2023, 95, e28209. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef]
- Kaur, T.; Dumoga, S.; Koul, V.; Singh, N. Modulating Neutrophil Extracellular Traps for Wound Healing. Biomater. Sci. 2020, 8, 3212–3223. [Google Scholar] [CrossRef] [PubMed]
- Stocker, M.; van Herk, W.; el Helou, S.; Dutta, S.; Schuerman, F.A.B.A.; van den Tooren-de Groot, R.K.; Wieringa, J.W.; Janota, J.; van der Meer-Kappelle, L.H.; Moonen, R.; et al. C-Reactive Protein, Procalcitonin, and White Blood Count to Rule Out Neonatal Early-Onset Sepsis Within 36 Hours: A Secondary Analysis of the Neonatal Procalcitonin Intervention Study. Clin. Infect. Dis. 2021, 73, e383–e390. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sadikot, R.T.; Adami, G.R.; Kalinichenko, V.V.; Pendyala, S.; Natarajan, V.; Zhao, Y.; Malik, A.B. FoxM1 Mediates the Progenitor Function of Type II Epithelial Cells in Repairing Alveolar Injury Induced by Pseudomonas Aeruginosa. J. Exp. Med. 2011, 208, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Rubio, I.; Osuchowski, M.F.; Shankar-Hari, M.; Skirecki, T.; Winkler, M.S.; Lachmann, G.; La Rosée, P.; Monneret, G.; Venet, F.; Bauer, M.; et al. Current Gaps in Sepsis Immunology: New Opportunities for Translational Research. Lancet Infect. Dis. 2019, 19, e422–e436. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.; Yu, Y.; Hong, Q.; Li, C.; Zhang, H.; Guo, K. Neutrophil Extracellular Traps Upregulate p21 and Suppress Cell Cycle Progression to Impair Endothelial Regeneration after Inflammatory Lung Injury. J. Clin. Med. 2024, 13, 1204. https://doi.org/10.3390/jcm13051204
Zhu S, Yu Y, Hong Q, Li C, Zhang H, Guo K. Neutrophil Extracellular Traps Upregulate p21 and Suppress Cell Cycle Progression to Impair Endothelial Regeneration after Inflammatory Lung Injury. Journal of Clinical Medicine. 2024; 13(5):1204. https://doi.org/10.3390/jcm13051204
Chicago/Turabian StyleZhu, Shuainan, Ying Yu, Qianya Hong, Chenning Li, Hao Zhang, and Kefang Guo. 2024. "Neutrophil Extracellular Traps Upregulate p21 and Suppress Cell Cycle Progression to Impair Endothelial Regeneration after Inflammatory Lung Injury" Journal of Clinical Medicine 13, no. 5: 1204. https://doi.org/10.3390/jcm13051204